Interview with Tamir Gonen about MicroED
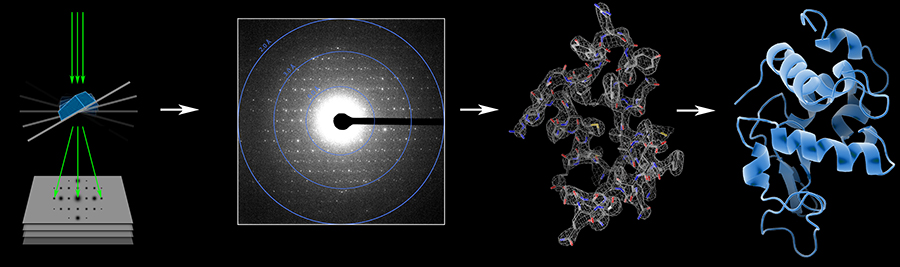
We demonstrated that it is feasible to determine high-resolution protein structures by electron crystallography of three-dimensional crystals in an electron cryo-microscope (cryo-EM). Lysozyme microcrystals were frozen on an electron microscopy grid, and electron diffraction data collected to 1.7 Å resolution. We developed a data collection protocol to collect a full-tilt series in electron diffraction to atomic resolution. A single tilt series contains up to 90 individual diffraction patterns collected from a single crystal with tilt angle increment of 0.1°–1° and a total accumulated electron dose less than 10 electrons per Å2. We indexed the data from three crystals and used them for structure determination of lysozyme by molecular replacement followed by crystallographic refinement to 2.9 Å resolution. This proof of principle paves the way for the implementation of a new technique, which we name MicroED, that may have wide applicability in structural biology.
In 2014 we further improved the MicroED method. Firstly, we developed an improved data collection protocol for MicroED called continuous rotation. Microcrystals are continuously rotated during data collection yielding improved data and allowing data processing with the crystallographic software tool MOSFLM, resulting in improved resolution for the model protein lysozyme to 2.5 Å resolution. These improvements pave the way for the broad implementation and application of MicroED in structural biology. Current efforts include new phasing methods, automation, and program development.
Secondly, we used the improved MicroED protocols for data collection and analysis to determine the structure of catalase. Bovine liver catalase crystals that were only ~160 nm thick were used for the structure analysis. A single crystal yielded data to 3.2 Å resolution enabling rapid structure determination.
In 2015 we published the first two previously unknown structures determined by MicroED. The structures of two peptides from the toxic core of α-synuclein of Parkinson’s disease. The structures were determined from vanishingly small crystals, only ~200 nm thick and wide, and yielded 1.4 Å resolution. These structures, which are currently the highest resolution structures determined to date by any cryo-EM method, show new and important structural information that could aid in the development of pharmaceuticals against this devastating neurological disease. The study, which was published by Nature, also show a number of protons for the very first time.
2021: Structures of two globular proteins were determined ab initio using microcrystal electron diffraction (MicroED) data. Microcrystals were identified using a scanning electron microscope (SEM) and thinned with a focused ion-beam (FIB) to produce crystalline lamellae of ideal thickness. Continuous rotation data were collected using an ultra-low exposure rate on a Falcon 4 direct electron detector in electron-counting mode. For the first sample, triclinic lysozyme extending to 0.87 Å resolution, an ideal helical fragment of only three alanine residues provided initial phases. These phases were improved using density modification, allowing the entire atomic structure to be built automatically. A similar approach was successful on a second macromolecular sample, proteinase K, which is much larger and diffracted to 1.5 Å resolution. These results demonstrate that macromolecules can be determined to sub-Ångström resolution by MicroED and that ab initio phasing can be successfully applied to counting data collected on a direct electron detector.
Follow MicroED on Twitter #MicroED
Raw datasets available for download
- Lysozyme: DOI 10.15785/SBGRID/222
- Catalase: DOI 10.15785/SBGRID/186
- α-synuclein G11A: DOI 10.15785/SBGRID/223
- GSNQNNF: DOI 10.15785/SBGRID/819
- VFAThiaGlu: DOI 10.15785/SBGRID/820
Hardware
The MicroED Stage Controller page contains a full description of the stage rotation controller developed at Janelia Research Campus. This device is not needed on Thermo Fisher systems from 2015 or later.
Software downloads
All software that we developed for MicroED data processing can be found on the downloads pages.
See also
Selected publications
Clabbers, Max T. B.; Martynowycz, Michael W.; Hattne, Johan; Gonen, Tamir
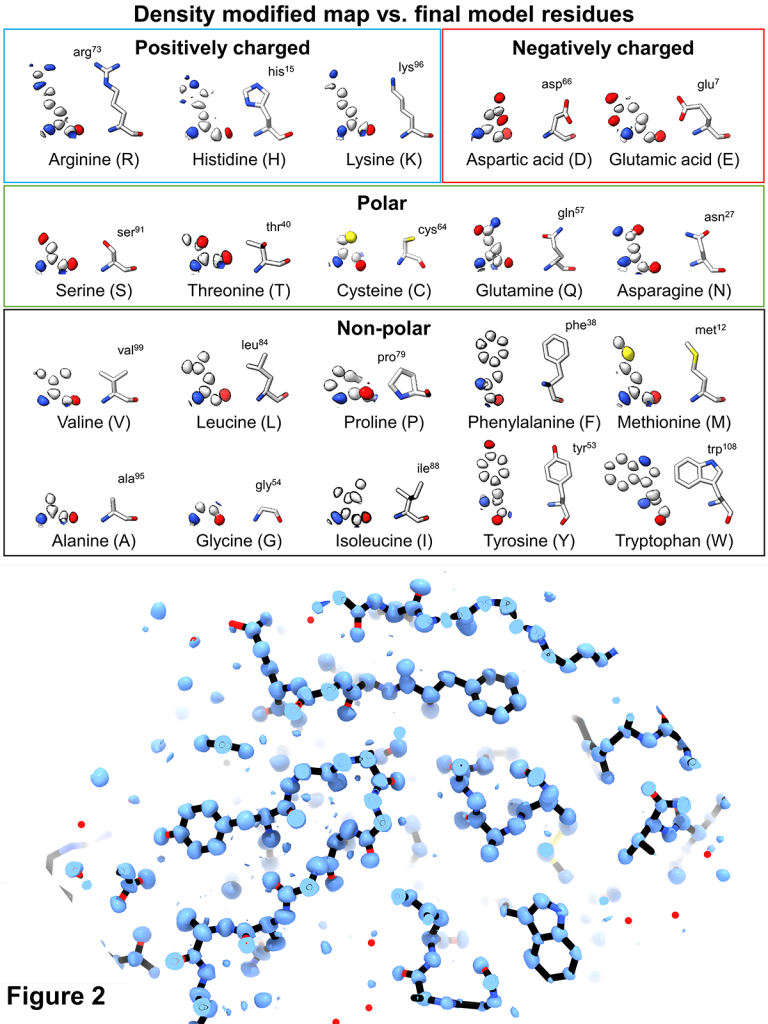
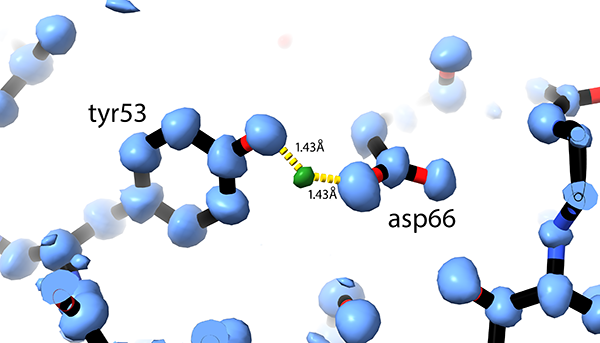
Hydrogens and hydrogen-bond networks in macromolecular MicroED data
In: J Struct Biol X, vol. 6, pp. 100078, 2022.
@article{nokey,
title = {Hydrogens and hydrogen-bond networks in macromolecular MicroED data},
author = {Max T.B. Clabbers and Michael W. Martynowycz and Johan Hattne and Tamir Gonen},
doi = {10.1016/j.yjsbx.2022.100078},
year = {2022},
date = {2022-11-10},
urldate = {2022-11-10},
journal = {J Struct Biol X},
volume = {6},
pages = {100078},
organization = {bioRxiv},
abstract = {Microcrystal electron diffraction (MicroED) is a powerful technique utilizing electron cryo-microscopy (cryo-EM) for protein structure determination of crystalline samples too small for X-ray crystallography. Electrons interact with the electrostatic potential of the sample, which means that the scattered electrons carry information about the charged state of atoms and provide relatively stronger contrast for visualizing hydrogen atoms. Accurately identifying the positions of hydrogen atoms, and by extension the hydrogen bonding networks, is of importance for understanding protein structure and function, in particular for drug discovery. However, identification of individual hydrogen atom positions typically requires atomic resolution data, and has thus far remained elusive for macromolecular MicroED. Recently, we presented the ab initio structure of triclinic hen egg-white lysozyme at 0.87 Å resolution. The corresponding data were recorded under low exposure conditions using an electron-counting detector from thin crystalline lamellae. Here, using these subatomic resolution MicroED data, we identified over a third of all hydrogen atom positions based on strong difference peaks, and directly visualize hydrogen bonding interactions and the charged states of residues. Furthermore, we find that the hydrogen bond lengths are more accurately described by the inter-nuclei distances than the centers of mass of the corresponding electron clouds. We anticipate that MicroED, coupled with ongoing advances in data collection and refinement, can open further avenues for structural biology by uncovering the hydrogen atoms and hydrogen bonding interactions underlying protein structure and function.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Clabbers, Max T. B.; Martynowycz, Michael W.; Hattne, Johan; Nannenga, Brent L.; Gonen, Tamir
Electron-counting MicroED data with the K2 and K3 direct electron detectors
In: J Struct Biol, vol. 214, iss. 4, 2022.
@article{pmid36044956,
title = {Electron-counting MicroED data with the K2 and K3 direct electron detectors},
author = {Max T.B. Clabbers and Michael W. Martynowycz and Johan Hattne and Brent L. Nannenga and Tamir Gonen},
doi = {10.1016/j.jsb.2022.107886},
year = {2022},
date = {2022-08-28},
urldate = {2022-08-28},
journal = {J Struct Biol},
volume = {214},
issue = {4},
organization = {bioRxiv},
abstract = {Microcrystal electron diffraction (MicroED) uses electron cryo-microscopy (cryo-EM) to collect diffraction data from small crystals during continuous rotation of the sample. As a result of advances in hardware as well as methods development, the data quality has continuously improved over the past decade, to the point where even macromolecular structures can be determined ab initio. Detectors suitable for electron diffraction should ideally have fast readout to record data in movie mode, and high sensitivity at low exposure rates to accurately report the intensities. Direct electron detectors are commonly used in cryo-EM imaging for their sensitivity and speed, but despite their availability are generally not used in diffraction. Primary concerns with diffraction experiments are the dynamic range and coincidence loss, which will corrupt the measurement if the flux exceeds the count rate of the detector. Here, we describe instrument setup and low-exposure MicroED data collection in electron-counting mode using K2 and K3 direct electron detectors and show that the integrated intensities can be effectively used to solve structures of two macromolecules between 1.2 Å and 2.8 Å. Even though a beam stop was not used in these studies we did not observe damage to the camera. As these cameras are already available in many cryo-EM facilities, this provides opportunities for users who do not have access to dedicated facilities for MicroED.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Martynowycz, Michael W; Clabbers, Max T B; Hattne, Johan; Gonen, Tamir
Ab initio phasing macromolecular structures using electron-counted MicroED data
In: Nat Methods, vol. 19, iss. 6, pp. 724–729, 2022, ISSN: 1548-7105.
@article{pmid35637302,
title = {Ab initio phasing macromolecular structures using electron-counted MicroED data},
author = {Michael W Martynowycz and Max T B Clabbers and Johan Hattne and Tamir Gonen},
doi = {10.1038/s41592-022-01485-4},
issn = {1548-7105},
year = {2022},
date = {2022-05-30},
urldate = {2022-05-01},
journal = {Nat Methods},
volume = {19},
issue = {6},
pages = {724--729},
abstract = {Structures of two globular proteins were determined ab initio using microcrystal electron diffraction (MicroED) data that were collected on a direct electron detector in counting mode. Microcrystals were identified using a scanning electron microscope (SEM) and thinned with a focused ion beam (FIB) to produce crystalline lamellae of ideal thickness. Continuous-rotation data were collected using an ultra-low exposure rate to enable electron counting in diffraction. For the first sample, triclinic lysozyme extending to a resolution of 0.87 Å, an ideal helical fragment of only three alanine residues provided initial phases. These phases were improved using density modification, allowing the entire atomic structure to be built automatically. A similar approach was successful on a second macromolecular sample, proteinase K, which is much larger and diffracted to a resolution of 1.5 Å. These results demonstrate that macromolecules can be determined to sub-ångström resolution by MicroED and that ab initio phasing can be successfully applied to counting data.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Martynowycz, Michael W; Clabbers, Max T B; Unge, Johan; Hattne, Johan; Gonen, Tamir
Benchmarking the ideal sample thickness in cryo-EM
In: Proc Natl Acad Sci U S A, vol. 118, no. 49, 2021, ISSN: 1091-6490.
@article{pmid34873060,
title = {Benchmarking the ideal sample thickness in cryo-EM},
author = {Michael W Martynowycz and Max T B Clabbers and Johan Unge and Johan Hattne and Tamir Gonen},
doi = {10.1073/pnas.2108884118},
issn = {1091-6490},
year = {2021},
date = {2021-12-01},
urldate = {2021-12-01},
journal = {Proc Natl Acad Sci U S A},
volume = {118},
number = {49},
abstract = {The relationship between sample thickness and quality of data obtained is investigated by microcrystal electron diffraction (MicroED). Several electron microscopy (EM) grids containing proteinase K microcrystals of similar sizes from the same crystallization batch were prepared. Each grid was transferred into a focused ion beam and a scanning electron microscope in which the crystals were then systematically thinned into lamellae between 95- and 1,650-nm thick. MicroED data were collected at either 120-, 200-, or 300-kV accelerating voltages. Lamellae thicknesses were expressed in multiples of the corresponding inelastic mean free path to allow the results from different acceleration voltages to be compared. The quality of the data and subsequently determined structures were assessed using standard crystallographic measures. Structures were reliably determined with similar quality from crystalline lamellae up to twice the inelastic mean free path. Lower resolution diffraction was observed at three times the mean free path for all three accelerating voltages, but the data quality was insufficient to yield structures. Finally, no coherent diffraction was observed from lamellae thicker than four times the calculated inelastic mean free path. This study benchmarks the ideal specimen thickness with implications for all cryo-EM methods.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Zhu, Lan; Bu, Guanhong; Jing, Liang; Shi, Dan; Lee, Ming-Yue; Gonen, Tamir; Liu, Wei; Nannenga, Brent L.
Structure Determination from Lipidic Cubic Phase Embedded Microcrystals by MicroED
In: Structure, vol. 28, no. 10, pp. 1149–1159.e4, 2020.
@article{2020_Zhu,
title = {Structure Determination from Lipidic Cubic Phase Embedded Microcrystals by MicroED},
author = {Lan Zhu and Guanhong Bu and Liang Jing and Dan Shi and Ming-Yue Lee and Tamir Gonen and Wei Liu and Brent L. Nannenga},
url = {https://cryoem.ucla.edu/wp-content/uploads/2020_Zhu.pdf, Main text},
doi = {https://doi.org/10.1016/j.str.2020.07.006},
year = {2020},
date = {2020-10-06},
journal = {Structure},
volume = {28},
number = {10},
pages = {1149--1159.e4},
abstract = {The lipidic cubic phase (LCP) technique has proved to facilitate the growth of high-quality crystals that are otherwise difficult to grow by other methods. However, the crystal size optimization process could be time and resource consuming, if it ever happens. Therefore, improved techniques for structure determination using these small crystals is an important strategy in diffraction technology development. Microcrystal electron diffraction (MicroED) is a technique that uses a cryo-transmission electron microscopy to collect electron diffraction data and determine high-resolution structures from very thin micro- and nanocrystals. In this work, we have used modified LCP and MicroED protocols to analyze crystals embedded in LCP converted by 2-methyl-2,4-pentanediol or lipase, including Proteinase K crystals grown in solution, cholesterol crystals, and human adenosine A2A receptor crystals grown in LCP. These results set the stage for the use of MicroED to analyze microcrystalline samples grown in LCP, especially for those highly challenging membrane protein targets.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Jones, Christopher G; Martynowycz, Michael W; Hattne, Johan; Fulton, Tyler J; Stoltz, Brian M; Rodriguez, Jose A; Nelson, Hosea M; Gonen, Tamir
The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination
In: ACS Cent Sci, vol. 4, no. 11, pp. 1587–1592, 2018.
@article{pmid30555912,
title = {The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination},
author = {Christopher G Jones and Michael W Martynowycz and Johan Hattne and Tyler J Fulton and Brian M Stoltz and Jose A Rodriguez and Hosea M Nelson and Tamir Gonen},
doi = {10.1021/acscentsci.8b00760},
year = {2018},
date = {2018-11-02},
journal = {ACS Cent Sci},
volume = {4},
number = {11},
pages = {1587--1592},
abstract = {In the many scientific endeavors that are driven by organic chemistry, unambiguous identification of small molecules is of paramount importance. Over the past 50 years, NMR and other powerful spectroscopic techniques have been developed to address this challenge. While almost all of these techniques rely on inference of connectivity, the unambiguous determination of a small molecule's structure requires X-ray and/or neutron diffraction studies. In practice, however, X-ray crystallography is rarely applied in routine organic chemistry due to intrinsic limitations of both the analytes and the technique. Here we report the use of the electron cryo-microscopy (cryoEM) method microcrystal electron diffraction (MicroED) to provide routine and unambiguous structural determination of small organic molecules. From simple powders, with minimal sample preparation, we could collect high-quality MicroED data from nanocrystals (∼100 nm, ∼10-15 g) resulting in atomic resolution (<1 Å) crystal structures in minutes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Liu, Shian; Gonen, Tamir
MicroED structure of the NaK ion channel reveals a Na⁺ partition process into the selectivity filter
In: Commun Biol, vol. 1, pp. 38, 2018.
@article{pmid30167468,
title = {MicroED structure of the NaK ion channel reveals a Na⁺ partition process into the selectivity filter},
author = {Shian Liu and Tamir Gonen},
doi = {10.1038/s42003-018-0040-8},
year = {2018},
date = {2018-05-03},
journal = {Commun Biol},
volume = {1},
pages = {38},
abstract = {Sodium (Na+) is a ubiquitous and important inorganic salt mediating many critical biological processes such as neuronal excitation, signaling, and facilitation of various transporters. The hydration states of Na+ are proposed to play critical roles in determining the conductance and the selectivity of Na+ channels, yet they are rarely captured by conventional structural biology means. Here we use the emerging cryo-electron microscopy (cryoEM) method micro-electron diffraction (MicroED) to study the structure of a prototypical tetrameric Na+-conducting channel, NaK, to 2.5 Å resolution from nano-crystals. Two new conformations at the external site of NaK are identified, allowing us to visualize a partially hydrated Na+ ion at the entrance of the channel pore. A process of dilation coupled with Na+ movement is identified leading to valuable insights into the mechanism of ion conduction and gating. This study lays the ground work for future studies using MicroED in membrane protein biophysics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hattne, Johan; Shi, Dan; Glynn, Calina; Zee, Chih-Te; Gallagher-Jones, Marcus; Martynowycz, Michael W; Rodriguez, Jose A; Gonen, Tamir
Analysis of Global and Site-Specific Radiation Damage in Cryo-EM
In: Structure, vol. 26, no. 5, pp. 759–766, 2018.
@article{pmid29706530,
title = {Analysis of Global and Site-Specific Radiation Damage in Cryo-EM},
author = {Johan Hattne and Dan Shi and Calina Glynn and Chih-Te Zee and Marcus Gallagher-Jones and Michael W Martynowycz and Jose A Rodriguez and Tamir Gonen},
doi = {10.1016/j.str.2018.03.021},
year = {2018},
date = {2018-05-01},
journal = {Structure},
volume = {26},
number = {5},
pages = {759--766},
abstract = {Micro-crystal electron diffraction (MicroED) combines the efficiency of electron scattering with diffraction to allow structure determination from nano-sized crystalline samples in cryoelectron microscopy (cryo-EM). It has been used to solve structures of a diverse set of biomolecules and materials, in some cases to sub-atomic resolution. However, little is known about the damaging effects of the electron beam on samples during such measurements. We assess global and site-specific damage from electron radiation on nanocrystals of proteinase K and of a prion hepta-peptide and find that the dynamics of electron-induced damage follow well-established trends observed in X-ray crystallography. Metal ions are perturbed, disulfide bonds are broken, and acidic side chains are decarboxylated while the diffracted intensities decay exponentially with increasing exposure. A better understanding of radiation damage in MicroED improves our assessment and processing of all types of cryo-EM data.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
de la Cruz, Jason M; Hattne, Johan; Shi, Dan; Seidler, Paul; Rodriguez, Jose; Reyes, Francis E; Sawaya, Michael R; Cascio, Duilio; Weiss, Simon C; Kim, Sun Kyung; Hinck, Cynthia S; Hinck, Andrew P; Calero, Guillermo; Eisenberg, David; Gonen, Tamir
Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED
In: Nat. Methods, vol. 14, no. 4, pp. 399–402, 2017.
@article{pmid28192420,
title = {Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED},
author = {Jason M de la Cruz and Johan Hattne and Dan Shi and Paul Seidler and Jose Rodriguez and Francis E Reyes and Michael R Sawaya and Duilio Cascio and Simon C Weiss and Sun Kyung Kim and Cynthia S Hinck and Andrew P Hinck and Guillermo Calero and David Eisenberg and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2017_delacruz_a.pdf, Main text},
doi = {10.1038/nmeth.4178},
year = {2017},
date = {2017-02-13},
journal = {Nat. Methods},
volume = {14},
number = {4},
pages = {399--402},
abstract = {Traditionally, crystallographic analysis of macromolecules has depended on large, well-ordered crystals, which often require significant effort to obtain. Even sizable crystals sometimes suffer from pathologies that render them inappropriate for high-resolution structure determination. Here we show that fragmentation of large, imperfect crystals into microcrystals or nanocrystals can provide a simple path for high-resolution structure determination by the cryoEM method MicroED and potentially by serial femtosecond crystallography.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hattne, Johan; Shi, Dan; de la Cruz, Jason M; Reyes, Francis E; Gonen, Tamir
Modeling truncated pixel values of faint reflections in MicroED images
In: J Appl Crystallogr, vol. 49, no. Pt 3, pp. 1029–1034, 2016.
@article{pmid27275145,
title = {Modeling truncated pixel values of faint reflections in MicroED images},
author = {Johan Hattne and Dan Shi and Jason M de la Cruz and Francis E Reyes and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/hattne_2016.pdf, Main text},
doi = {10.1107/S1600576716007196},
year = {2016},
date = {2016-06-01},
journal = {J Appl Crystallogr},
volume = {49},
number = {Pt 3},
pages = {1029--1034},
abstract = {The weak pixel counts surrounding the Bragg spots in a diffraction image are important for establishing a model of the background underneath the peak and estimating the reliability of the integrated intensities. Under certain circumstances, particularly with equipment not optimized for low-intensity measurements, these pixel values may be corrupted by corrections applied to the raw image. This can lead to truncation of low pixel counts, resulting in anomalies in the integrated Bragg intensities, such as systematically higher signal-to-noise ratios. A correction for this effect can be approximated by a three-parameter lognormal distribution fitted to the weakly positive-valued pixels at similar scattering angles. The procedure is validated by the improved refinement of an atomic model against structure factor amplitudes derived from corrected micro-electron diffraction (MicroED) images.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Shi, Dan; Nannenga, Brent L; de la Cruz, Jason M; Liu, Jinyang; Sawtelle, Steven; Calero, Guillermo; Reyes, Francis E; Hattne, Johan; Gonen, Tamir
The collection of MicroED data for macromolecular crystallography
In: Nat Protoc, vol. 11, no. 5, pp. 895–904, 2016.
@article{pmid27077331,
title = {The collection of MicroED data for macromolecular crystallography},
author = {Dan Shi and Brent L Nannenga and Jason M de la Cruz and Jinyang Liu and Steven Sawtelle and Guillermo Calero and Francis E Reyes and Johan Hattne and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2016_shi.pdf, Main text},
doi = {10.1038/nprot.2016.046},
year = {2016},
date = {2016-04-14},
journal = {Nat Protoc},
volume = {11},
number = {5},
pages = {895--904},
abstract = {The formation of large, well-ordered crystals for crystallographic experiments remains a crucial bottleneck to the structural understanding of many important biological systems. To help alleviate this problem in crystallography, we have developed the MicroED method for the collection of electron diffraction data from 3D microcrystals and nanocrystals of radiation-sensitive biological material. In this approach, liquid solutions containing protein microcrystals are deposited on carbon-coated electron microscopy grids and are vitrified by plunging them into liquid ethane. MicroED data are collected for each selected crystal using cryo-electron microscopy, in which the crystal is diffracted using very few electrons as the stage is continuously rotated. This protocol gives advice on how to identify microcrystals by light microscopy or by negative-stain electron microscopy in samples obtained from standard protein crystallization experiments. The protocol also includes information about custom-designed equipment for controlling crystal rotation and software for recording experimental parameters in diffraction image metadata. Identifying microcrystals, preparing samples and setting up the microscope for diffraction data collection take approximately half an hour for each step. Screening microcrystals for quality diffraction takes roughly an hour, and the collection of a single data set is ∼10 min in duration. Complete data sets and resulting high-resolution structures can be obtained from a single crystal or by merging data from multiple crystals.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Rodriguez, Jose A; Ivanova, Magdalena I; Sawaya, Michael R; Cascio, Duilio; Reyes, Francis E; Shi, Dan; Sangwan, Smriti; Guenther, Elizabeth L; Johnson, Lisa M; Zhang, Meng; Jiang, Lin; Arbing, Mark A; Nannenga, Brent L; Hattne, Johan; Whitelegge, Julian; Brewster, Aaron S; Messerschmidt, Marc; Boutet, Sébastien; Sauter, Nicholas K; Gonen, Tamir; Eisenberg, David S
Structure of the toxic core of α-synuclein from invisible crystals
In: Nature, vol. 525, no. 7570, pp. 486–490, 2015.
@article{pmid26352473,
title = {Structure of the toxic core of α-synuclein from invisible crystals},
author = {Jose A Rodriguez and Magdalena I Ivanova and Michael R Sawaya and Duilio Cascio and Francis E Reyes and Dan Shi and Smriti Sangwan and Elizabeth L Guenther and Lisa M Johnson and Meng Zhang and Lin Jiang and Mark A Arbing and Brent L Nannenga and Johan Hattne and Julian Whitelegge and Aaron S Brewster and Marc Messerschmidt and Sébastien Boutet and Nicholas K Sauter and Tamir Gonen and David S Eisenberg},
url = {https://cryoem.ucla.edu/wp-content/uploads/2015rodriguez.pdf, Main text},
doi = {10.1038/nature15368},
year = {2015},
date = {2015-09-09},
journal = {Nature},
volume = {525},
number = {7570},
pages = {486--490},
abstract = {The protein α-synuclein is the main component of Lewy bodies, the neuron-associated aggregates seen in Parkinson disease and other neurodegenerative pathologies. An 11-residue segment, which we term NACore, appears to be responsible for amyloid formation and cytotoxicity of human α-synuclein. Here we describe crystals of NACore that have dimensions smaller than the wavelength of visible light and thus are invisible by optical microscopy. As the crystals are thousands of times too small for structure determination by synchrotron X-ray diffraction, we use micro-electron diffraction to determine the structure at atomic resolution. The 1.4 Å resolution structure demonstrates that this method can determine previously unknown protein structures and here yields, to our knowledge, the highest resolution achieved by any cryo-electron microscopy method to date. The structure exhibits protofibrils built of pairs of face-to-face β-sheets. X-ray fibre diffraction patterns show the similarity of NACore to toxic fibrils of full-length α-synuclein. The NACore structure, together with that of a second segment, inspires a model for most of the ordered portion of the toxic, full-length α-synuclein fibril, presenting opportunities for the design of inhibitors of α-synuclein fibrils.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hattne, Johan; Reyes, Francis E; Nannenga, Brent L; Shi, Dan; de la Cruz, Jason M; Leslie, Andrew G W; Gonen, Tamir
MicroED data collection and processing
In: Acta Crystallogr A Found Adv, vol. 71, no. Pt 4, pp. 353–360, 2015.
@article{pmid26131894,
title = {MicroED data collection and processing},
author = {Johan Hattne and Francis E Reyes and Brent L Nannenga and Dan Shi and Jason M de la Cruz and Andrew G W Leslie and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2015_hattne.pdf, Main text},
doi = {10.1107/S2053273315010669},
year = {2015},
date = {2015-07-01},
journal = {Acta Crystallogr A Found Adv},
volume = {71},
number = {Pt 4},
pages = {353--360},
abstract = {MicroED, a method at the intersection of X-ray crystallography and electron cryo-microscopy, has rapidly progressed by exploiting advances in both fields and has already been successfully employed to determine the atomic structures of several proteins from sub-micron-sized, three-dimensional crystals. A major limiting factor in X-ray crystallography is the requirement for large and well ordered crystals. By permitting electron diffraction patterns to be collected from much smaller crystals, or even single well ordered domains of large crystals composed of several small mosaic blocks, MicroED has the potential to overcome the limiting size requirement and enable structural studies on difficult-to-crystallize samples. This communication details the steps for sample preparation, data collection and reduction necessary to obtain refined, high-resolution, three-dimensional models by MicroED, and presents some of its unique challenges.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nannenga, Brent L; Shi, Dan; Hattne, Johan; Reyes, Francis E; Gonen, Tamir
Structure of catalase determined by MicroED
In: Elife, vol. 3, pp. e03600, 2014.
@article{pmid25303172,
title = {Structure of catalase determined by MicroED},
author = {Brent L Nannenga and Dan Shi and Johan Hattne and Francis E Reyes and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2014_nannenga_b.pdf, Main text},
doi = {10.7554/eLife.03600},
year = {2014},
date = {2014-10-10},
journal = {Elife},
volume = {3},
pages = {e03600},
abstract = {MicroED is a recently developed method that uses electron diffraction for structure determination from very small three-dimensional crystals of biological material. Previously we used a series of still diffraction patterns to determine the structure of lysozyme at 2.9 Å resolution with MicroED (Shi et al., 2013). Here we present the structure of bovine liver catalase determined from a single crystal at 3.2 Å resolution by MicroED. The data were collected by continuous rotation of the sample under constant exposure and were processed and refined using standard programs for X-ray crystallography. The ability of MicroED to determine the structure of bovine liver catalase, a protein that has long resisted atomic analysis by traditional electron crystallography, demonstrates the potential of this method for structure determination.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nannenga, Brent L; Shi, Dan; Leslie, Andrew G W; Gonen, Tamir
High-resolution structure determination by continuous-rotation data collection in MicroED
In: Nat. Methods, vol. 11, no. 9, pp. 927–930, 2014.
@article{pmid25086503,
title = {High-resolution structure determination by continuous-rotation data collection in MicroED},
author = {Brent L Nannenga and Dan Shi and Andrew G W Leslie and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2014_nannenga.pdf, Main text},
doi = {10.1038/nmeth.3043},
year = {2014},
date = {2014-08-03},
journal = {Nat. Methods},
volume = {11},
number = {9},
pages = {927--930},
abstract = {MicroED uses very small three-dimensional protein crystals and electron diffraction for structure determination. We present an improved data collection protocol for MicroED called 'continuous rotation'. Microcrystals are continuously rotated during data collection, yielding more accurate data. The method enables data processing with the crystallographic software tool MOSFLM, which resulted in improved resolution for the model protein lysozyme. These improvements are paving the way for the broad implementation and application of MicroED in structural biology.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Iadanza, Matthew G; Gonen, Tamir
A suite of software for processing MicroED data of extremely small protein crystals
In: J Appl Crystallogr, vol. 47, no. Pt 3, pp. 1140–1145, 2014.
@article{pmid24904248,
title = {A suite of software for processing MicroED data of extremely small protein crystals},
author = {Matthew G Iadanza and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2014_iadanzagonen.pdf, Main text},
doi = {10.1107/S1600576714008073},
year = {2014},
date = {2014-06-01},
journal = {J Appl Crystallogr},
volume = {47},
number = {Pt 3},
pages = {1140--1145},
abstract = {Electron diffraction of extremely small three-dimensional crystals (MicroED) allows for structure determination from crystals orders of magnitude smaller than those used for X-ray crystallography. MicroED patterns, which are collected in a transmission electron microscope, were initially not amenable to indexing and intensity extraction by standard software, which necessitated the development of a suite of programs for data processing. The MicroED suite was developed to accomplish the tasks of unit-cell determination, indexing, background subtraction, intensity measurement and merging, resulting in data that can be carried forward to molecular replacement and structure determination. This ad hoc solution has been modified for more general use to provide a means for processing MicroED data until the technique can be fully implemented into existing crystallographic software packages. The suite is written in Python and the source code is available under a GNU General Public License.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Shi, Dan; Nannenga, Brent L; Iadanza, Matthew G; Gonen, Tamir
Three-dimensional electron crystallography of protein microcrystals
In: Elife, vol. 2, pp. e01345, 2013.
@article{pmid24252878,
title = {Three-dimensional electron crystallography of protein microcrystals},
author = {Dan Shi and Brent L Nannenga and Matthew G Iadanza and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2013_shi.pdf, Main text},
doi = {10.7554/eLife.01345},
year = {2013},
date = {2013-11-19},
journal = {Elife},
volume = {2},
pages = {e01345},
abstract = {We demonstrate that it is feasible to determine high-resolution protein structures by electron crystallography of three-dimensional crystals in an electron cryo-microscope (CryoEM). Lysozyme microcrystals were frozen on an electron microscopy grid, and electron diffraction data collected to 1.7 Å resolution. We developed a data collection protocol to collect a full-tilt series in electron diffraction to atomic resolution. A single tilt series contains up to 90 individual diffraction patterns collected from a single crystal with tilt angle increment of 0.1-1° and a total accumulated electron dose less than 10 electrons per angstrom squared. We indexed the data from three crystals and used them for structure determination of lysozyme by molecular replacement followed by crystallographic refinement to 2.9 Å resolution. This proof of principle paves the way for the implementation of a new technique, which we name 'MicroED', that may have wide applicability in structural biology. DOI: http://dx.doi.org/10.7554/eLife.01345.001.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wisedchaisri, Goragot; Gonen, Tamir
In: Structure, vol. 19, no. 7, pp. 976–987, 2011.
@article{pmid21742264,
title = {Fragment-Based Phase Extension for Three-Dimensional Structure Determination of Membrane Proteins by Electron Crystallography},
author = {Goragot Wisedchaisri and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2011_wisedchaisri_1.pdf, Main text},
doi = {10.1016/j.str.2011.04.008},
year = {2011},
date = {2011-07-13},
journal = {Structure},
volume = {19},
number = {7},
pages = {976--987},
abstract = {In electron crystallography, membrane protein structure is determined from two-dimensional crystals where the protein is embedded in a membrane. Once large and well-ordered 2D crystals are grown, one of the bottlenecks in electron crystallography is the collection of image data to directly provide experimental phases to high resolution. Here, we describe an approach to bypass this bottleneck, eliminating the need for high-resolution imaging. We use the strengths of electron crystallography in rapidly obtaining accurate experimental phase information from low-resolution images and accurate high-resolution amplitude information from electron diffraction. The low-resolution experimental phases were used for the placement of α helix fragments and extended to high resolution using phases from the fragments. Phases were further improved by density modifications followed by fragment expansion and structure refinement against the high-resolution diffraction data. Using this approach, structures of three membrane proteins were determined rapidly and accurately to atomic resolution without high-resolution image data.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Relevant reviews and book chapters
Clark, Lisa J.; Bu, Guanhong; Nannenga, Brent L.; Gonen, Tamir
MicroED for the study of protein–ligand interactions and the potential for drug discovery
In: Nat Rev Chem, vol. 5, pp. 853–858, 2021.
@article{Clark2021,
title = {MicroED for the study of protein–ligand interactions and the potential for drug discovery},
author = {Lisa J. Clark and Guanhong Bu and Brent L. Nannenga and Tamir Gonen},
doi = {10.1038/s41570-021-00332-y},
year = {2021},
date = {2021-10-27},
urldate = {2021-10-27},
journal = {Nat Rev Chem},
volume = {5},
pages = {853–858},
abstract = {Microcrystal electron diffraction (MicroED) is an electron cryo-microscopy (cryo-EM) technique used to determine molecular structures with crystals that are a millionth the size needed for traditional single-crystal X-ray crystallography. An exciting use of MicroED is in drug discovery and development, where it can be applied to the study of proteins and small molecule interactions, and for structure determination of natural products. The structures are then used for rational drug design and optimization. In this Perspective, we discuss the current applications of MicroED for structure determination of protein–ligand complexes and potential future applications in drug discovery.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Mu, Xuelang; Gillman, Cody; Nguyen, Chi; Gonen, Tamir
An Overview of Microcrystal Electron Diffraction (MicroED)
In: Annu Rev Biochem, vol. 90, pp. 431–450, 2021, ISSN: 1545-4509.
@article{pmid34153215,
title = {An Overview of Microcrystal Electron Diffraction (MicroED)},
author = {Xuelang Mu and Cody Gillman and Chi Nguyen and Tamir Gonen},
doi = {10.1146/annurev-biochem-081720-020121},
issn = {1545-4509},
year = {2021},
date = {2021-06-20},
urldate = {2021-06-00},
journal = {Annu Rev Biochem},
volume = {90},
pages = {431--450},
abstract = {The bedrock of drug discovery and a key tool for understanding cellular function and drug mechanisms of action is the structure determination of chemical compounds, peptides, and proteins. The development of new structure characterization tools, particularly those that fill critical gaps in existing methods, presents important steps forward for structural biology and drug discovery. The emergence of microcrystal electron diffraction (MicroED) expands the application of cryo-electron microscopy to include samples ranging from small molecules and membrane proteins to even large protein complexes using crystals that are one-billionth the size of those required for X-ray crystallography. This review outlines the conception, achievements, and exciting future trajectories for MicroED, an important addition to the existing biophysical toolkit.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Danelius, E; Gonen, T
Protein and Small Molecule Structure Determination by the Cryo-EM Method MicroED
In: Owens, Raymond J. (Ed.): vol. 2305, pp. 323–342, 2021.
@inbook{pmid33950397,
title = {Protein and Small Molecule Structure Determination by the Cryo-EM Method MicroED},
author = {E Danelius and T Gonen},
editor = {Raymond J. Owens},
doi = {10.1007/978-1-0716-1406-8_16},
year = {2021},
date = {2021-05-06},
journal = {Methods Mol Biol},
volume = {2305},
pages = {323--342},
series = {Methods in Molecular Biology},
abstract = {Microcrystal Electron Diffraction (MicroED) is the newest cryo-electron microscopy (cryo-EM) method, with over 70 protein, peptide, and several small organic molecule structures already determined. In MicroED, micro- or nanocrystalline samples in solution are deposited on electron microscopy grids and examined in a cryo-electron microscope, ideally under cryogenic conditions. Continuous rotation diffraction data are collected and then processed using conventional X-ray crystallography programs. The protocol outlined here details how to obtain and identify the nanocrystals, how to set up the microscope for screening and for MicroED data collection, and how to collect and process data to complete high-resolution structures. For well-behaving crystals with high-resolution diffraction in cryo-EM, the entire process can be achieved in less than an hour.},
keywords = {},
pubstate = {published},
tppubtype = {inbook}
}
Nannenga, Brent L; Gonen, Tamir
The cryo-EM method microcrystal electron diffraction (MicroED)
In: Nat. Methods, vol. 16, no. 5, pp. 369–379, 2019.
@article{pmid31040436,
title = {The cryo-EM method microcrystal electron diffraction (MicroED)},
author = {Brent L Nannenga and Tamir Gonen},
doi = {10.1038/s41592-019-0395-x},
year = {2019},
date = {2019-04-29},
journal = {Nat. Methods},
volume = {16},
number = {5},
pages = {369--379},
abstract = {In 2013 we established a cryo-electron microscopy (cryo-EM) technique called microcrystal electron diffraction (MicroED). Since that time, data collection and analysis schemes have been fine-tuned, and structures for more than 40 different proteins, oligopeptides and organic molecules have been determined. Here we review the MicroED technique and place it in context with other structure-determination methods. We showcase example structures solved by MicroED and provide practical advice to prospective users.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Martynowycz, Michael W; Gonen, Tamir
From electron crystallography of 2D crystals to MicroED of 3D crystals
In: Curr Opin Colloid Interface Sci, vol. 34, pp. 9–16, 2018.
@article{pmid30166936,
title = {From electron crystallography of 2D crystals to MicroED of 3D crystals},
author = {Michael W Martynowycz and Tamir Gonen},
doi = {10.1016/j.cocis.2018.01.010},
year = {2018},
date = {2018-03-01},
journal = {Curr Opin Colloid Interface Sci},
volume = {34},
pages = {9--16},
abstract = {Electron crystallography is widespread in material science applications, but for biological samples its use has been restricted to a handful of examples where two-dimensional (2D) crystals or helical samples were studied either by electron diffraction and/or imaging. Electron crystallography in cryoEM, was developed in the mid-1970s and used to solve the structure of several membrane proteins and some soluble proteins. In 2013, a new method for cryoEM was unveiled and named Micro-crystal Electron Diffraction, or MicroED, which is essentially three-dimensional (3D) electron crystallography of microscopic crystals. This method uses truly 3D crystals, that are about a billion times smaller than those typically used for X-ray crystallography, for electron diffraction studies. There are several important differences and some similarities between electron crystallography of 2D crystals and MicroED. In this review, we describe the development of these techniques, their similarities and differences, and offer our opinion of future directions in both fields.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nannenga, Brent L; Gonen, Tamir
Protein structure determination by MicroED
In: Curr. Opin. Struct. Biol., vol. 27, pp. 24–31, 2014.
@article{pmid24709395,
title = {Protein structure determination by MicroED},
author = {Brent L Nannenga and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2014_nannengagonen.pdf, Main text},
doi = {10.1016/j.sbi.2014.03.004},
year = {2014},
date = {2014-08-01},
journal = {Curr. Opin. Struct. Biol.},
volume = {27},
pages = {24--31},
abstract = {In this review we discuss the current advances relating to structure determination from protein microcrystals with special emphasis on the newly developed method called MicroED. This method uses a transmission electron cryo-microscope to collect electron diffraction data from extremely small 3-dimensional (3D) crystals. MicroED has been used to solve the 3D structure of the model protein lysozyme to 2.9Å resolution. As the method further matures, MicroED promises to offer a unique and widely applicable approach to protein crystallography using nanocrystals.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nannenga, Brent L; Iadanza, Matthew G; Vollmar, Breanna S; Gonen, Tamir
In: Curr Protoc Protein Sci, vol. 72, no. 1, pp. 17.15.1–17.15.11, 2013.
@article{pmid23546618,
title = {Overview of Electron Crystallography of Membrane Proteins: Crystallization and Screening Strategies Using Negative Stain Electron Microscopy},
author = {Brent L Nannenga and Matthew G Iadanza and Breanna S Vollmar and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/nannenga_2013.pdf, Main text},
doi = {10.1002/0471140864.ps1715s72},
year = {2013},
date = {2013-04-01},
journal = {Curr Protoc Protein Sci},
volume = {72},
number = {1},
pages = {17.15.1--17.15.11},
chapter = {17},
abstract = {Electron cryomicroscopy, or cryoEM, is an emerging technique for studying the three-dimensional structures of proteins and large macromolecular machines. Electron crystallography is a branch of cryoEM in which structures of proteins can be studied at resolutions that rival those achieved by X-ray crystallography. Electron crystallography employs two-dimensional crystals of a membrane protein embedded within a lipid bilayer. The key to a successful electron crystallographic experiment is the crystallization, or reconstitution, of the protein of interest. This unit describes ways in which protein can be expressed, purified, and reconstituted into well-ordered two-dimensional crystals. A protocol is also provided for negative stain electron microscopy as a tool for screening crystallization trials. When large and well-ordered crystals are obtained, the structures of both protein and its surrounding membrane can be determined to atomic resolution.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wisedchaisri, Goragot; Gonen, Tamir
In: Electron Crystallography of Soluble and Membrane Proteins, vol. 955, Chapter 14, pp. 243–272, 2012.
@inbook{pmid23132065,
title = {Phasing Electron Diffraction Data by Molecular Replacement: Strategy for Structure Determination and Refinement},
author = {Goragot Wisedchaisri and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2013_wisedchaisrigonen.pdf, Main text},
doi = {10.1007/978-1-62703-176-9_14},
year = {2012},
date = {2012-10-07},
booktitle = {Electron Crystallography of Soluble and Membrane Proteins},
journal = {Methods Mol. Biol.},
volume = {955},
pages = {243--272},
chapter = {14},
abstract = {Electron crystallography is arguably the only electron cryomicroscopy (cryo EM) technique able to deliver atomic resolution data (better then 3 Å) for membrane proteins embedded in a membrane. The progress in hardware improvements and sample preparation for diffraction analysis resulted in a number of recent examples where increasingly higher resolutions were achieved. Other chapters in this book detail the improvements in hardware and delve into the intricate art of sample preparation for microscopy and electron diffraction data collection and processing. In this chapter, we describe in detail the protocols for molecular replacement for electron diffraction studies. The use of a search model for phasing electron diffraction data essentially eliminates the need of acquiring image data rendering it immune to aberrations from drift and charging effects that effectively lower the attainable resolution.},
keywords = {},
pubstate = {published},
tppubtype = {inbook}
}
Gonen, Tamir
The Collection of High-Resolution Electron Diffraction Data
In: Electron Crystallography of Soluble and Membrane Proteins, vol. 955, Chapter 9, pp. 153–169, 2012.
@inbook{pmid23132060,
title = {The Collection of High-Resolution Electron Diffraction Data},
author = {Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2013_gonen.pdf, Main text},
doi = {10.1007/978-1-62703-176-9_9},
year = {2012},
date = {2012-10-07},
booktitle = {Electron Crystallography of Soluble and Membrane Proteins},
journal = {Methods Mol. Biol.},
volume = {955},
pages = {153--169},
chapter = {9},
abstract = {A number of atomic-resolution structures of membrane proteins (better than 3Å resolution) have been determined recently by electron crystallography. While this technique was established more than 40 years ago, it is still in its infancy with regard to the two-dimensional (2D) crystallization, data collection, data analysis, and protein structure determination. In terms of data collection, electron crystallography encompasses both image acquisition and electron diffraction data collection. Other chapters in this volume outline protocols for image collection and analysis. This chapter, however, outlines detailed protocols for data collection by electron diffraction. These include microscope setup, electron diffraction data collection, and troubleshooting.},
keywords = {},
pubstate = {published},
tppubtype = {inbook}
}
Stokes, David L; Ubarretxena-Belandia, Iban; Gonen, Tamir; Engel, Andreas
High-Throughput Methods for Electron Crystallography
In: Electron Crystallography of Soluble and Membrane Proteins, vol. 955, Chapter 15, pp. 273–296, 2012.
@inbook{pmid23132066,
title = {High-Throughput Methods for Electron Crystallography},
author = {David L Stokes and Iban Ubarretxena-Belandia and Tamir Gonen and Andreas Engel},
url = {https://cryoem.ucla.edu/wp-content/uploads/2013_stokes.pdf, Main text},
doi = {10.1007/978-1-62703-176-9_15},
year = {2012},
date = {2012-10-07},
booktitle = {Electron Crystallography of Soluble and Membrane Proteins},
journal = {Methods Mol. Biol.},
volume = {955},
pages = {273--296},
chapter = {15},
abstract = {Membrane proteins play a tremendously important role in cell physiology and serve as a target for an increasing number of drugs. Structural information is key to understanding their function and for developing new strategies for combating disease. However, the complex physical chemistry associated with membrane proteins has made them more difficult to study than their soluble cousins. Electron crystallography has historically been a successful method for solving membrane protein structures and has the advantage of providing a native lipid environment for these proteins. Specifically, when membrane proteins form two-dimensional arrays within a lipid bilayer, electron microscopy can be used to collect images and diffraction and the corresponding data can be combined to produce a three-dimensional reconstruction, which under favorable conditions can extend to atomic resolution. Like X-ray crystallography, the quality of the structures are very much dependent on the order and size of the crystals. However, unlike X-ray crystallography, high-throughput methods for screening crystallization trials for electron crystallography are not in general use. In this chapter, we describe two alternative methods for high-throughput screening of membrane protein crystallization within the lipid bilayer. The first method relies on the conventional use of dialysis for removing detergent and thus reconstituting the bilayer; an array of dialysis wells in the standard 96-well format allows the use of a liquid-handling robot and greatly increases throughput. The second method relies on titration of cyclodextrin as a chelating agent for detergent; a specialized pipetting robot has been designed not only to add cyclodextrin in a systematic way, but to use light scattering to monitor the reconstitution process. In addition, the use of liquid-handling robots for making negatively stained grids and methods for automatically imaging samples in the electron microscope are described.},
keywords = {},
pubstate = {published},
tppubtype = {inbook}
}
Wisedchaisri, Goragot; Reichow, Steve L; Gonen, Tamir
Advances in Structural and Functional Analysis of Membrane Proteins by Electron Crystallography
In: Structure, vol. 19, no. 10, pp. 1381–1393, 2011.
@article{pmid22000511,
title = {Advances in Structural and Functional Analysis of Membrane Proteins by Electron Crystallography},
author = {Goragot Wisedchaisri and Steve L Reichow and Tamir Gonen},
url = {https://cryoem.ucla.edu/wp-content/uploads/2011_wisedchaisri.pdf, Main text},
doi = {10.1016/j.str.2011.09.001},
year = {2011},
date = {2011-10-12},
journal = {Structure},
volume = {19},
number = {10},
pages = {1381--1393},
abstract = {Electron crystallography is a powerful technique for the study of membrane protein structure and function in the lipid environment. When well-ordered two-dimensional crystals are obtained the structure of both protein and lipid can be determined and lipid-protein interactions analyzed. Protons and ionic charges can be visualized by electron crystallography and the protein of interest can be captured for structural analysis in a variety of physiologically distinct states. This review highlights the strengths of electron crystallography and the momentum that is building up in automation and the development of high throughput tools and methods for structural and functional analysis of membrane proteins by electron crystallography.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}