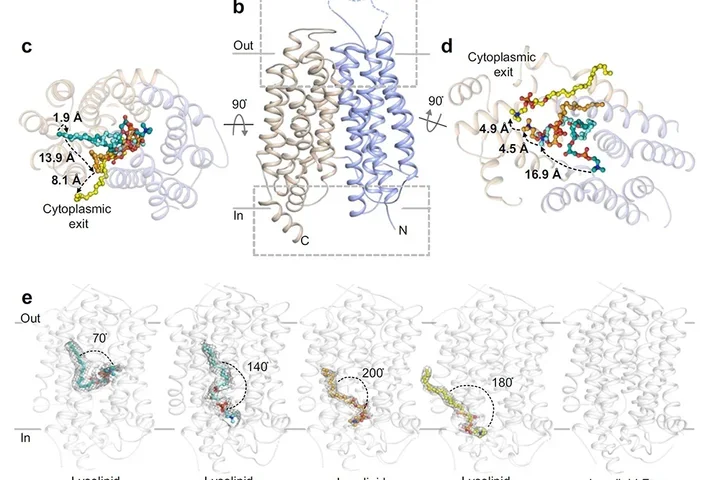
Breadcrumb Home Research Research Amino Acid Sensing and Transport Lipid Flipping Water Permeation Membrane Protein Complexes Membrane Transporters and Cellular Homeostasis Fragment‐Based Phase Extension MicroED Self‐Assembling Proteins Electrophysiology Vibrio cholera toxin secretion channel Yeast Kinetochore
Amino Acid Sensing and Transport Lipid Flipping Water Permeation Membrane Protein Complexes Membrane Transporters and Cellular Homeostasis Fragment‐Based Phase Extension MicroED Self‐Assembling Proteins Electrophysiology Vibrio cholera toxin secretion channel Yeast Kinetochore