
Lysosomal amino acid transceptor SLC38A9 and mTORC pathway activation: Recent advances in intracellular amino acid transport and mechanistic target of rapamycin complex 1 (mTORC1) signaling shed light on the solute carrier 38 family A member 9 (SLC38A9), a lysosomal transporter responsible for binding and translocation of several essential amino acids. Here we present the first crystal structure of SLC38A9 from Danio rerio in complex more…

Lipid flipping: Mfsd2a is the transporter for docosahexaenoic acid (DHA), an omega-3 fatty acid, across the blood brain barrier (BBB). Defects in Mfsd2a are linked to ailments from behavioral and motor dysfunctions to microcephaly. Mfsd2a transports long-chain unsaturated fatty-acids, including DHA and α-linolenic acid (ALA), that are attached to the zwitterionic lysophosphatidylcholine (LPC) headgroup. Even with the more…
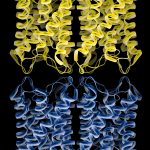
The dynamic regulation of water channels: Water channels, or aquaporins, form specialized channels in membranes for water permeation. These are extremely efficient channels that allow millions of water molecules to permeate the pore per second. Because they are channels, the cell can regulate their activity dynamically to help maintain homeostasis. In the case of the eye lens water channel aquaporin-0 (AQP0), more…

Membrane protein complexes: Our structure of the AQP0/CaM complex is the first for any full-length membrane channel in complex with this ubiquitous secondary messenger. Current efforts in the laboratory are to understand how Ca²⁺/CaM binds to and modulates the activity of other channels such as ion channels. We are also trying to understand more about the AQP-AKAP system, more…

Membrane transporters involved in nutrient uptake: Sugar uptake The major facilitator superfamily of membrane proteins is the largest collection of structurally related membrane proteins that transport a wide array of substrates. The proton-coupled sugar transporter XylE is the first member of the MFS that has been captured and structurally characterized in multiple transporting conformations including both the outward and inward facing more…
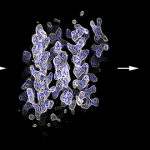
Fragment‐based phase extension: In electron crystallography membrane protein structure is determined from two-dimensional crystals where the protein is embedded in a membrane. Once large and well-ordered 2D crystals are grown one of the bottlenecks in electron crystallography is the collection of image data to directly provide experimental phases to high resolution. We developed a new approach to bypass more…
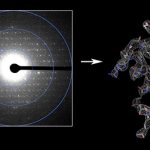
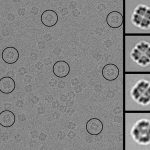
MicroED —Three‐dimensional electron crystallography of protein microcrystals: Interview with Tamir Gonen about MicroED We demonstrated that it is feasible to determine high-resolution protein structures by electron crystallography of three-dimensional crystals in an electron cryo-microscope (cryo-EM). Lysozyme microcrystals were frozen on an electron microscopy grid, and electron diffraction data collected to 1.7 Å resolution. We developed a data collection protocol to collect a full-tilt more…
Computational design of genetically encoded self‐assembling proteins: In collaboration with David Baker (HHMI, UW) we are designing genetically encoded self assembling proteins for cellular microcircuitry. We describe a general computational method for designing proteins that self-assemble to a desired symmetric architecture. Protein building blocks are docked together symmetrically to identify complementary packing arrangements, and low-energy protein-protein interfaces are then designed between the more…
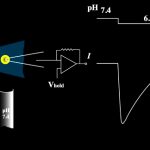
Electrophysiology—channel recordings toward structure‐function analysis: We use electrophysiology and patch clamping techniques to study the function of channels and transporters. We use the Xenopus oocyte expression system as well as whole cell patch but we also plan to record channel function from highly ordered two-dimensional crystals for a direct correlation between structure and function of target proteins as they are more…
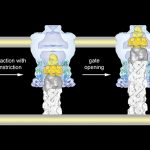
Structure of the vibrio cholera toxin secretion channel: In collaboration with Wim Hol (UW) we studied the structure of the vibrio cholera toxin secretion channel. The type II secretion system (T2SS) is a macromolecular complex spanning the bacterial inner and outer membranes of Gram-negative bacteria, including many pathogenic bacteria such as Vibrio cholerae and enterotoxigenic Escherichia coli. The T2SS secretes folded proteins including more…